写在前面——自从上次做了初级的题目之后,就一直在看这个中级的题目。因为中间有事耽搁了许久,所以间隔了很多天才做完。虽然按照视频和百度磕磕绊绊的把这个题目写完了,但是脑子还是一团浆糊。知道代码是干嘛的,但是不知道为什么要这么做。革命尚未成功,同志仍需努力…
题目原文:http://www.bio-info-trainee.com/3750.html
视频教程:https://www.bilibili.com/video/BV1cs411j75B?p=13
优质答案:https://www.jianshu.com/p/e15ee2cd3174
注意:如果library(…)报错的话,是因为没有安装包,需要install.packegs(…)安装对应的包。
1. 请根据R包org.Hs.eg.db找到下面ensembl 基因ID 对应的基因名(symbol)。
ENSG00000000003.13
ENSG00000000005.5
ENSG00000000419.11
ENSG00000000457.12
ENSG00000000460.15
ENSG00000000938.11
# 清空Environment中的变量,也可以直接点小扫帚清空
rm(list = ls())
# 避免把字符串项当成因子
options(stringsAsFactors = FALSE)
a <- read.table("practice/e1.txt")
# 思路:先得到egSYMBOL和egENSEMBL数据框
library(org.Hs.eg.db)
g2s <- toTable(org.Hs.egSYMBOL)
g2e <- toTable(org.Hs.egENSEMBL)
# 保留a中的v1小数点前面的部分,并将其赋给a的ensembl_id
a$ensembl_id <- unlist(lapply(a$V1, function(x){
strsplit(x, "[.]")[[1]][1]
}))
# 把a和g2e通过ensembl融合
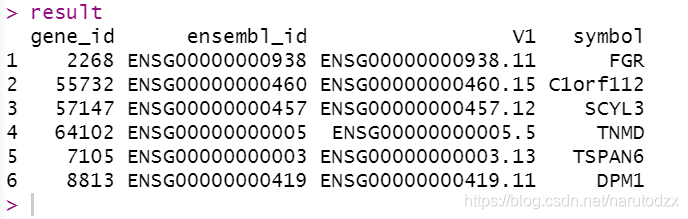
tmp <- merge(a, g2e, by = "ensembl_id")
# 最后再根据gene_id进行融合
result <- merge(tmp, g2s, by = "gene_id")

2. 根据R包hgu133a.db找到下面探针对应的基因名(symbol)。
1053_at
117_at
121_at
1255_g_at
1316_at
1320_at
1405_i_at
1431_at
1438_at
1487_at
1494_f_at
1598_g_at
160020_at
1729_at
177_at
rm(list = ls())
options(stringsAsFactors = FALSE)
b <- read.table("practice/e2.txt")
# 安装hgu133a.db包
# 若出错请查看 https://blog.csdn.net/narutodzx/article/details/119378949
# if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#
# BiocManager::install("hgu133a.db")
library(hgu133a.db)
ids <- toTable(hgu133aSYMBOL)
head(ids)
colnames(b) <- "probe_id"
result <- merge(ids, b, by="probe_id")

3. 找到R包CLL内置的数据集的表达矩阵里面的TP53基因的表达量,并且绘制在 progres.-stable分组的boxplot图。想想如何通过 ggpubr 进行美化。
rm(list = ls())
options(stringsAsFactors = FALSE)
# BiocManager::install("CLL")
# BiocManager::install("hgu95av2.db")
# 隐藏包在加载的时候显示的信息
suppressPackageStartupMessages(library(CLL))
# 加载数据
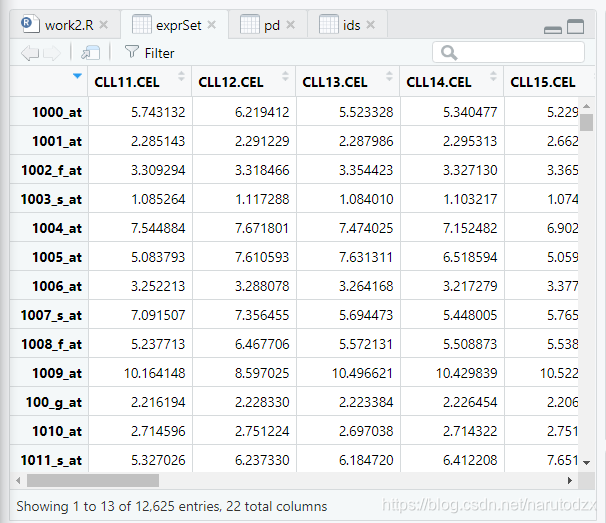
data(sCLLex)
sCLLex
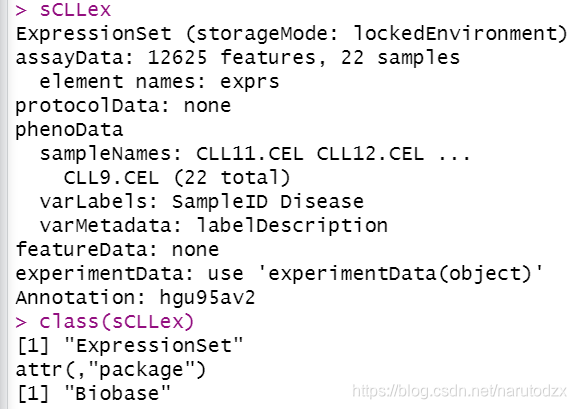
# 获得表达矩阵
# 用expr()提取assayData信息
exprSet <- exprs(sCLLex)
# 获得临床信息
# 用pData()提取phenoData信息
pd <- pData(sCLLex)
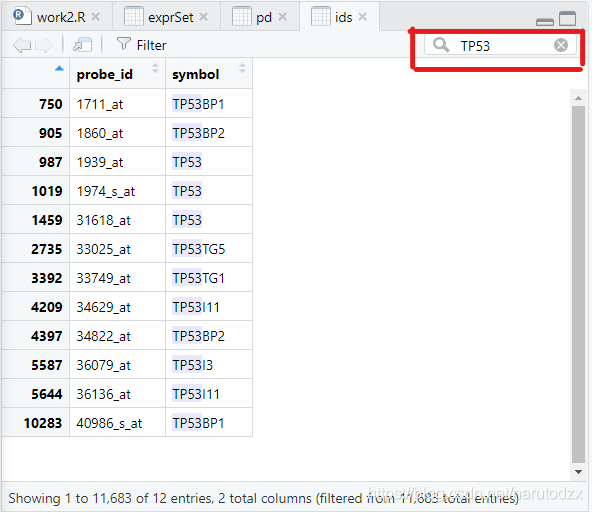
library(hgu95av2.db)
ids <- toTable(hgu95av2SYMBOL)
head(ids)
# 在ids中搜索TP53

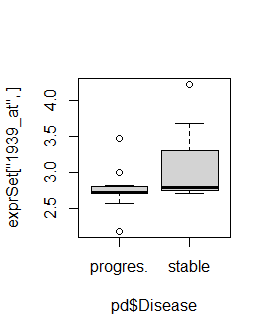
# 绘图
boxplot(exprSet['1939_at',] ~ pd$Disease) #signal
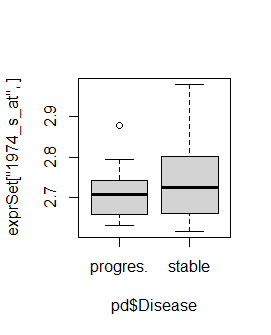
boxplot(exprSet['1974_s_at',] ~ pd$Disease)
boxplot(exprSet['31618_at',] ~ pd$Disease)
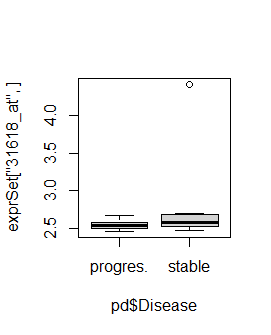
我看ggpubr中的ggboxplot函数都是对同一个数据框中的列进行绘图,不知道怎么对两个数据框中的列绘图,挖个坑,学会了再来填…
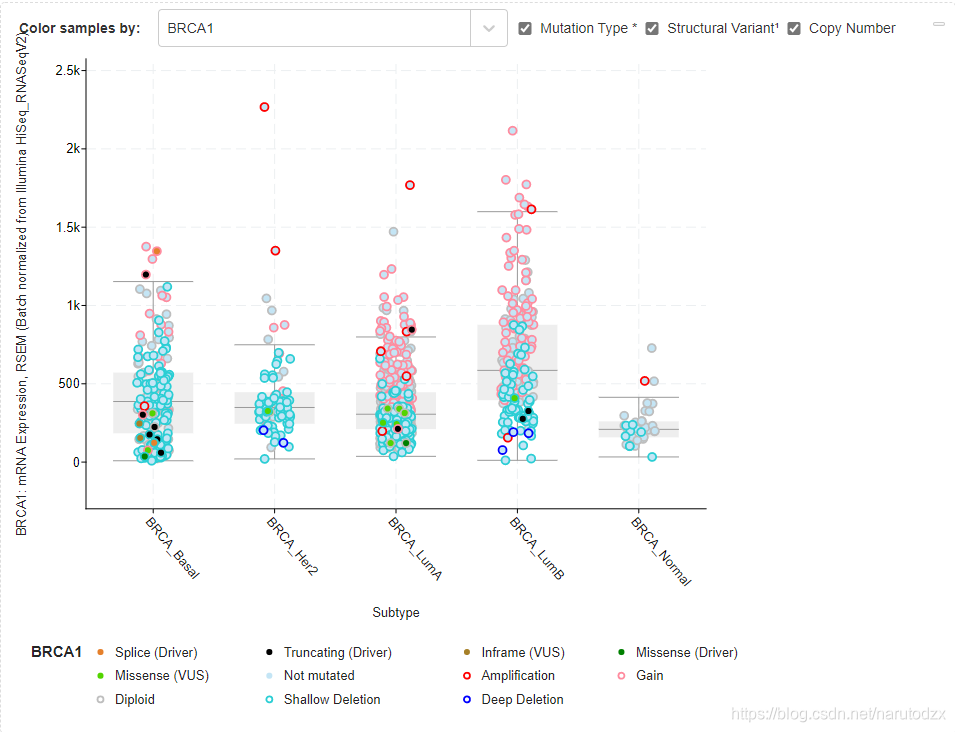
提示:使用http://www.cbioportal.org/index.do 定位数据集:http://www.cbioportal.org/datasets
表达情况:http://www.cbioportal.org/results/plots?cancer_study_list=brca_tcga_pan_can_atlas_2018&Z_SCORE_THRESHOLD=2.0&RPPA_SCORE_THRESHOLD=2.0&profileFilter=mutations%2Cfusion%2Cgistic&case_set_id=brca_tcga_pan_can_atlas_2018_cnaseq&gene_list=BRCA1&geneset_list=%20&tab_index=tab_visualize&Action=Submit&plots_horz_selection=%7B%22dataType%22%3A%22clinical_attribute%22%2C%22selectedDataSourceOption%22%3A%22SUBTYPE%22%7D&plots_vert_selection=%7B%22selectedGeneOption%22%3A672%7D&plots_coloring_selection=%7B%7D
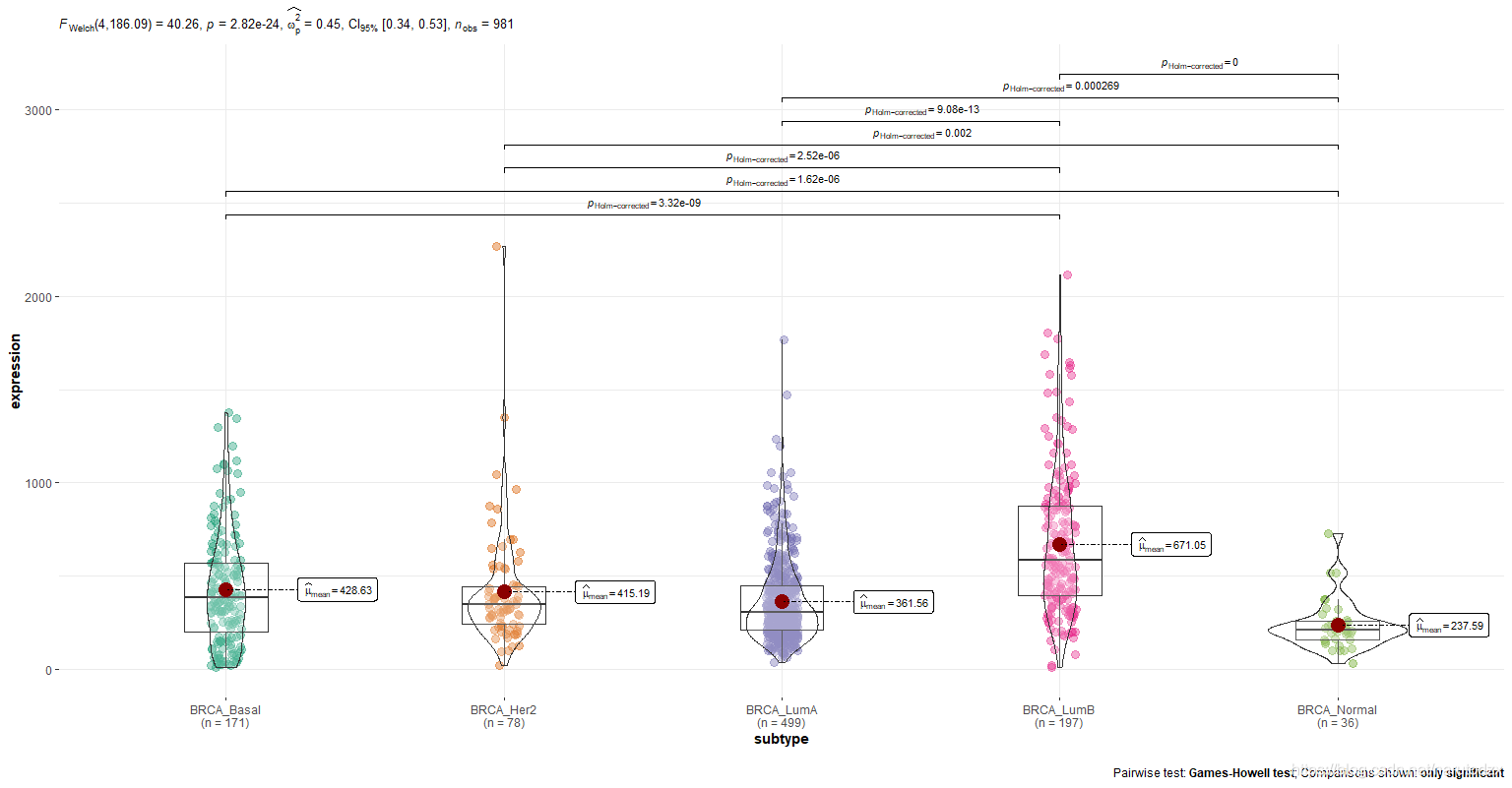
rm(list = ls())
options(stringsAsFactors = FALSE)
d <- read.table("practice/plot.txt", sep = "\t", fill = T, header = T)
# 列重新命名
colnames(d) <- c("id", "subtype", "expression", "mut", "cna")
# 绘图
# install.packages("ggstatsplot")
library(ggstatsplot)
ggbetweenstats(data = d, x = subtype, y = expression)
网站查询:http://www.oncolnc.org/kaplan/?lower=50&upper=50&cancer=BRCA&gene_id=7157&raw=TP53&species=mRNA
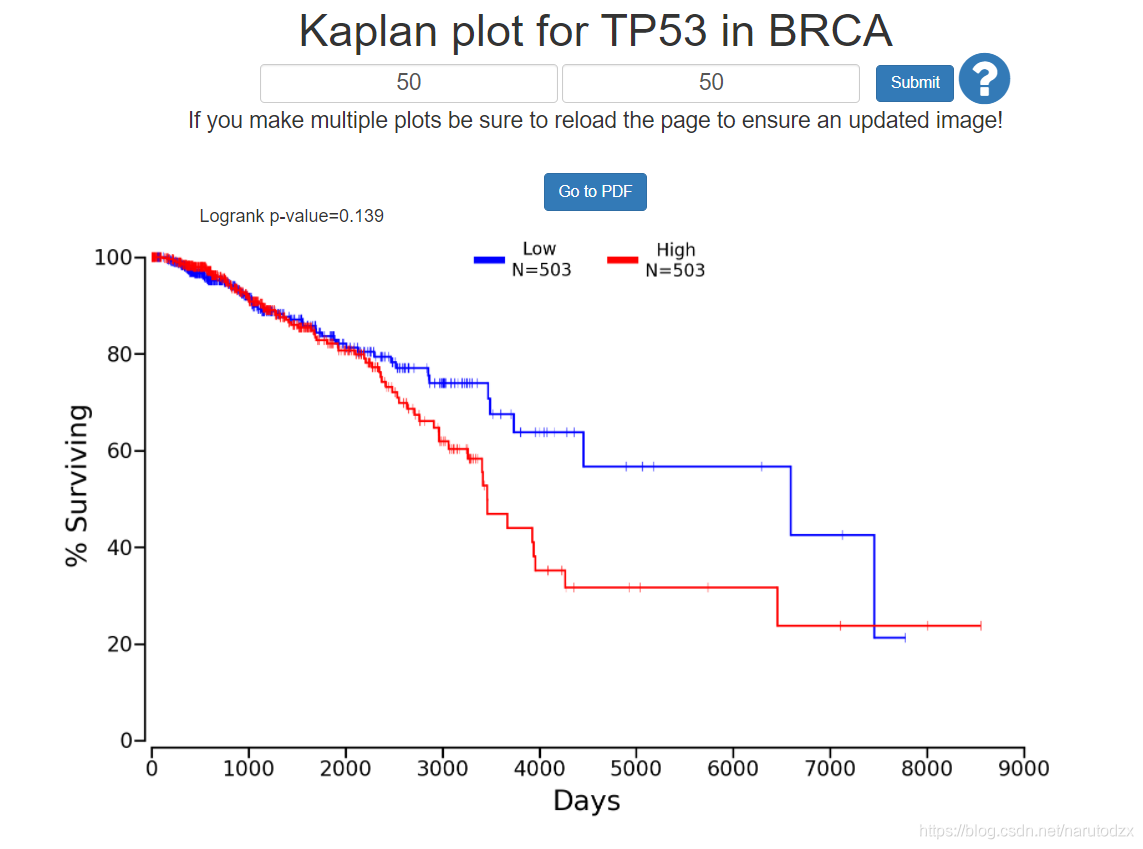
代码验证
rm(list = ls())
options(stringsAsFactors = FALSE)
e <- read.csv("practice/BRCA_7157_50_50.csv", header = T)
library(ggplot2)
library(survival)
library(survminer)
# table(e$Status)
e$Status <- ifelse(e$Status=='Dead',1,0)
sfit <- survfit(Surv(Days, Status)~Group, data=e)
sfit
summary(sfit)
# 绘制生存曲线
ggsurvplot(sfit, conf.int=F, pval=TRUE)
# ggsave('survival_TP53_in_BRCA_TCGA.png')
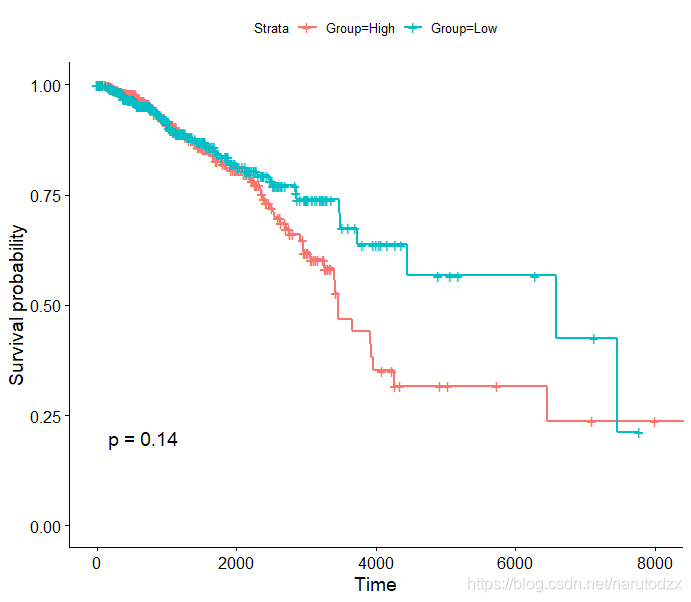
根据P值可以看出影响不大?
6. 下载数据集GSE17215的表达矩阵并且提取下面的基因画热图。
ACTR3B ANLN BAG1 BCL2 BIRC5 BLVRA CCNB1 CCNE1 CDC20 CDC6 CDCA1 CDH3 CENPF CEP55 CXXC5 EGFR ERBB2 ESR1 EXO1 FGFR4 FOXA1 FOXC1 GPR160 GRB7 KIF2C KNTC2 KRT14 KRT17 KRT5 MAPT MDM2 MELK MIA MKI67 MLPH MMP11 MYBL2 MYC NAT1 ORC6L PGR PHGDH PTTG1 RRM2 SFRP1 SLC39A6 TMEM45B TYMS UBE2C UBE2T
提示:根据基因名拿到探针ID,缩小表达矩阵绘制热图,没有检查到的基因直接忽略即可。
rm(list = ls())
options(stringsAsFactors = FALSE)
# 下载GEO数据集遇到网络问题的解决方法:https://www.jianshu.com/p/2686e257b250#comments
# 加载数据
library(GEOquery)
# ?getGEO
# 获取GSE17215_series_matrix.txt.gz
gset <- getGEO("GSE17215", destdir = ".", getGPL = F)
# 保存数据,方便以后使用
save(gset, file = "GSE17215_eSet.Rdata")
# 加载文件,注意文件所在位置
load('GSE17215_eSet.Rdata')
# 获得表达矩阵 方法同第3题
class(gset)
length(gset)
gset[[1]]
class(gset[[1]])
dat <- exprs(gset[[1]])
dim(dat)
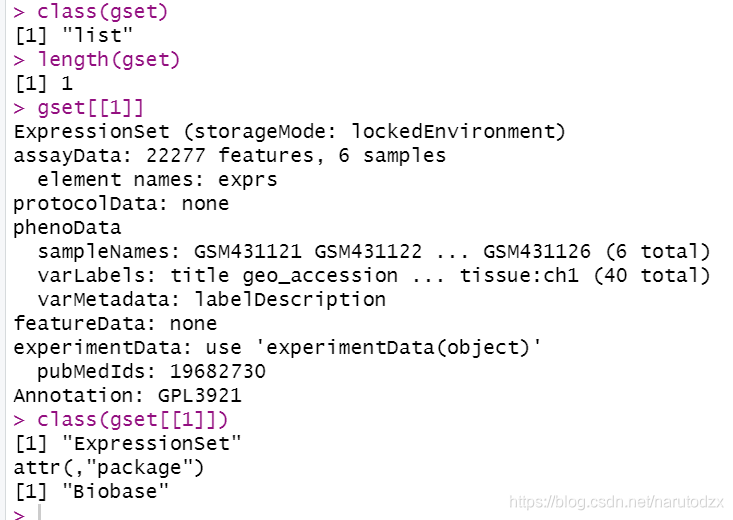
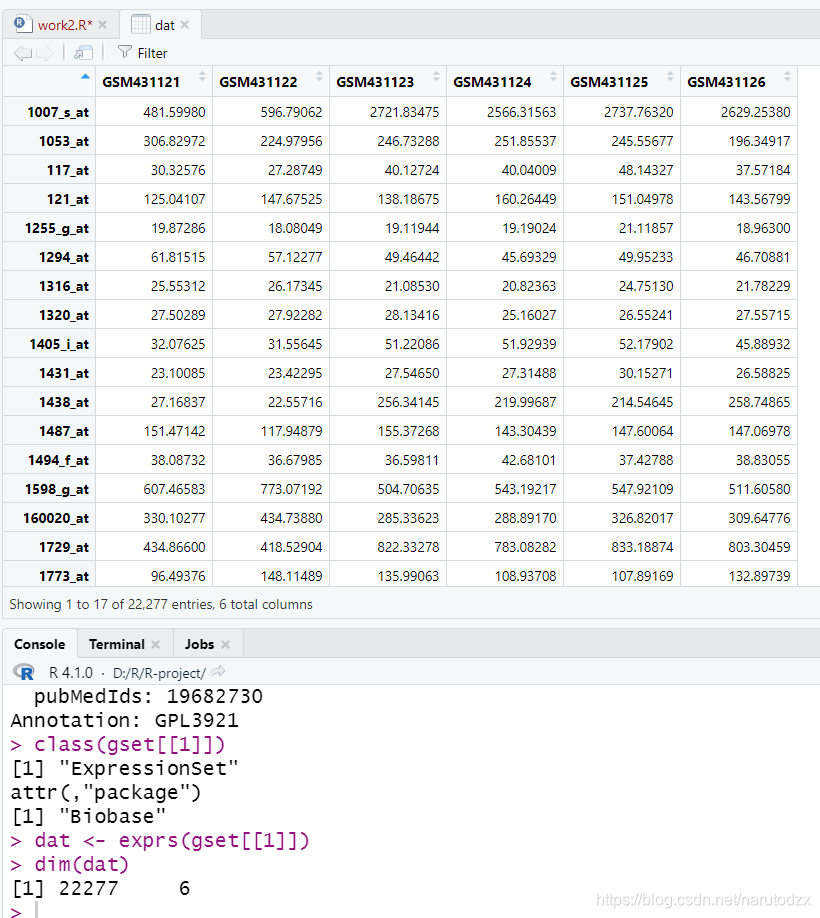
获得表达矩阵还有一种方法,见:如何成功getGEO https://www.jianshu.com/p/73580b051fa9?utm_campaign=maleskine…&utm_content=note&utm_medium=seo_notes&utm_source=recommendation
# 处理数据 同第2题
library(hgu133a.db)
ids <- toTable(hgu133aSYMBOL)
# head(ids)
dat <- dat[ids$probe_id,]
head(dat)
dim(dat)
# 对dat中的每行数据取中位数,赋值给ids中新建的median中
ids$median <- apply(dat,1,median)
# 首先对symbol进行排序,然后按照median排序(降序)
ids <- ids[order(ids$symbol,ids$median,decreasing = T),]
# 去除重复的gene ,保留每个基因最大表达量结果
ids <- ids[!duplicated(ids$symbol),]
# 新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
dat <- dat[ids$probe_id,]
# 把ids的symbol这一列中的行名作为dat的行名
rownames(dat) <- ids$symbol
head(dat)
dim(dat)
数据处理前: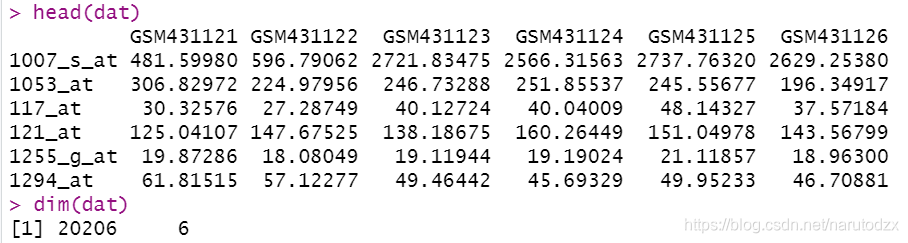
数据处理后: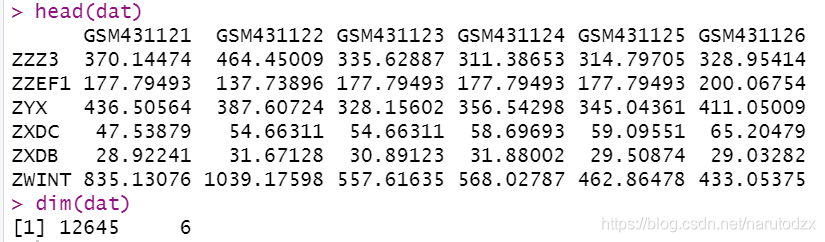
# 绘制热图
ng="ACTR3B ANLN BAG1 BCL2 BIRC5 BLVRA CCNB1 CCNE1 CDC20 CDC6 CDCA1 CDH3 CENPF CEP55 CXXC5 EGFR ERBB2 ESR1 EXO1 FGFR4 FOXA1 FOXC1 GPR160 GRB7 KIF2C KNTC2 KRT14 KRT17 KRT5 MAPT MDM2 MELK MIA MKI67 MLPH MMP11 MYBL2 MYC NAT1 ORC6L PGR PHGDH PTTG1 RRM2 SFRP1 SLC39A6 TMEM45B TYMS UBE2C UBE2T"
# 将字符串按空格分隔开,得到一个list,然后取list中的[[1]],得到分割后的字符串
ng=strsplit(ng,' ')[[1]]
ng
# 将ng中不存在的基因剔除
table(ng %in% rownames(dat))
ng=ng[ng %in% rownames(dat)]
table(ng %in% rownames(dat))
# 根据ng的值在dat中进行匹配
dat=dat[ng,]
# 绘图
dat=log2(dat)
pheatmap::pheatmap(dat,scale = 'row')

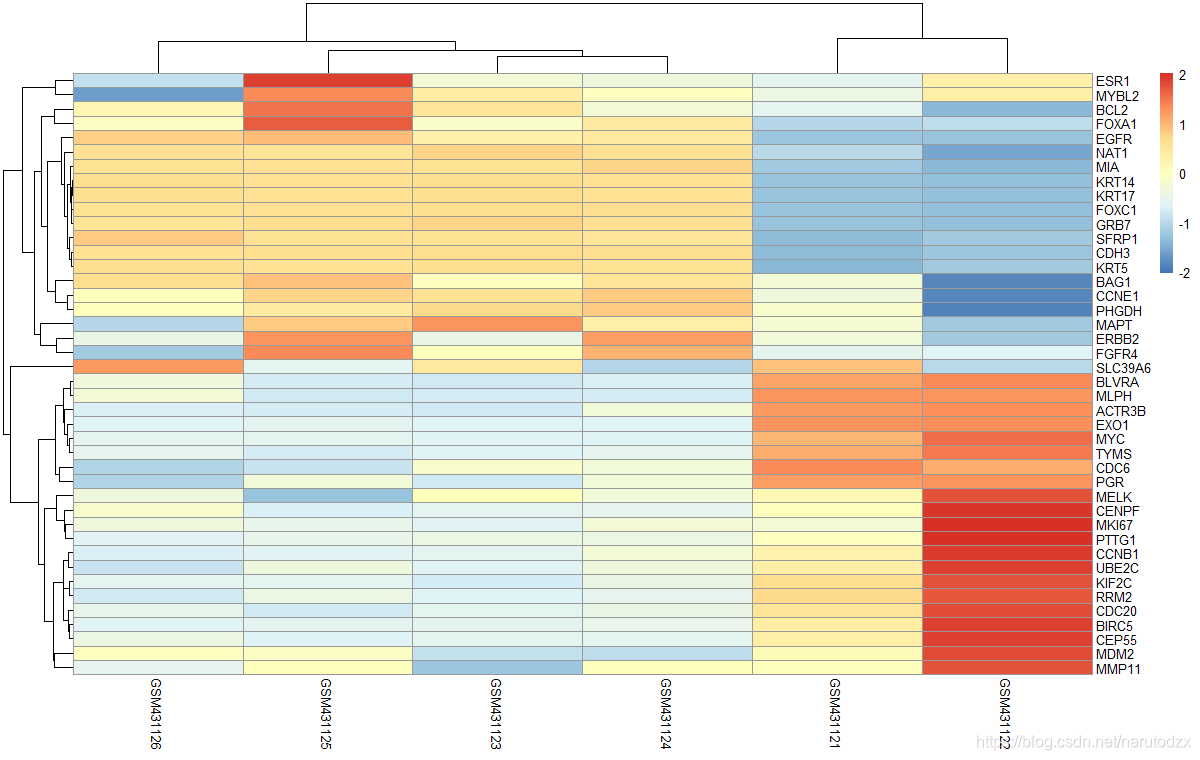
7. 下载数据集GSE24673的表达矩阵计算样本的相关性并且绘制热图,需要标记上样本分组信息。
rm(list = ls())
options(stringsAsFactors = FALSE)
# 下载并保存数据集
f='GSE24673_eSet.Rdata'
library(GEOquery)
# 设置网络,提高下载成功率
options( 'download.file.method.GEOquery' = 'libcurl' )
if(!file.exists(f)){
gset <- getGEO('GSE24673', destdir=".",
AnnotGPL = F,
getGPL = F)
save(gset,file=f)
}
load('GSE24673_eSet.Rdata')
# 表达矩阵
dat=exprs(gset[[1]])
# dim(dat)
# head(dat)
# 提取phenoData信息
pd=pData(gset[[1]])
# 根据pd数据中的第8列(source_name_ch1)进行分组
group_list=c('rbc','rbc','rbc',
'rbn','rbn','rbn',
'rbc','rbc','rbc',
'normal','normal')
# 计算样本的相关性并且绘制热图
M=cor(dat)
pheatmap::pheatmap(M)
# 标记样本分组信息
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(M)
pheatmap::pheatmap(M,annotation_col = tmp)
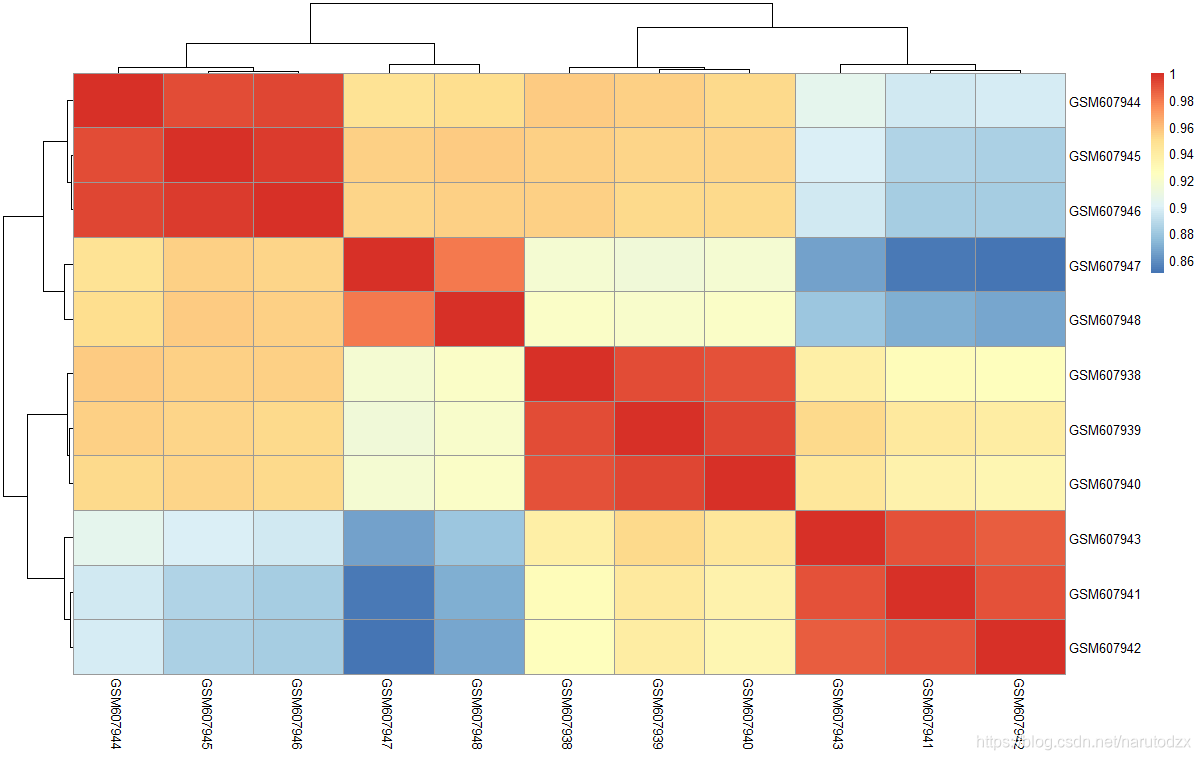
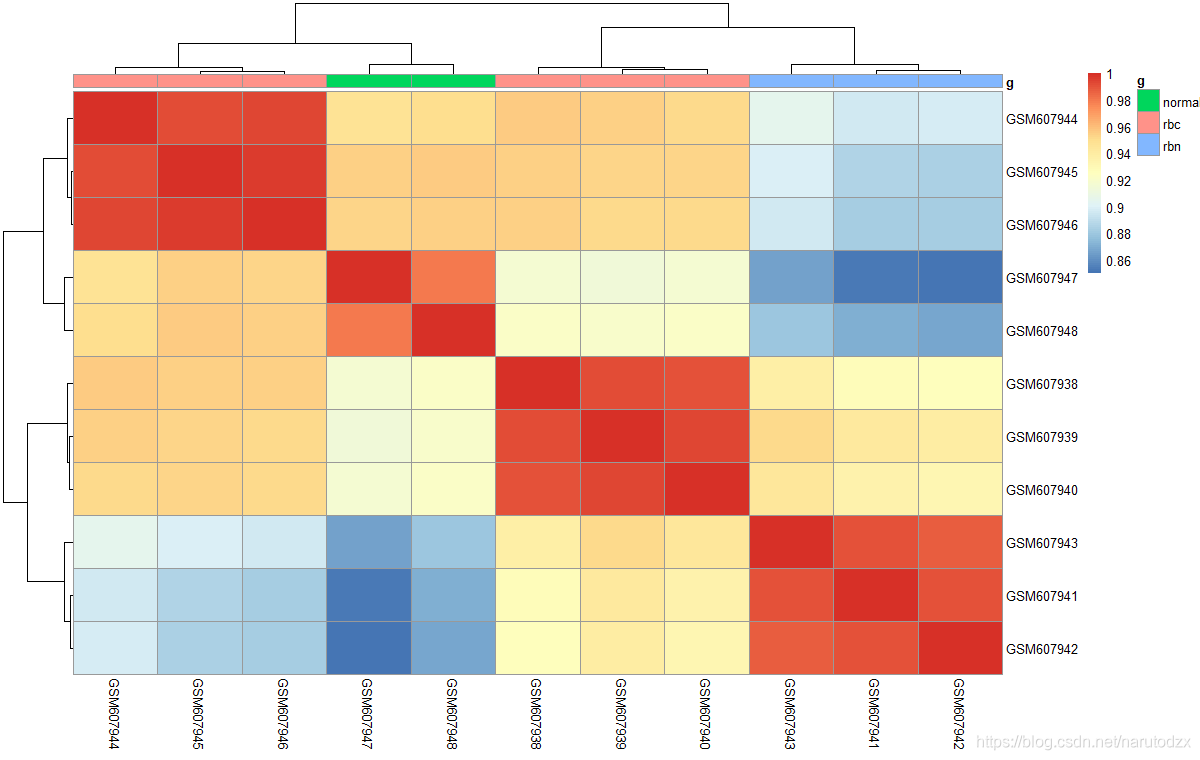
8. 找到 GPL6244 platform of Affymetrix Human Gene 1.0 ST Array 对应的R的bioconductor注释包,并且安装它。
如何查找对应的注释包:https://www.jianshu.com/p/73580b051fa9?utm_campaign=maleskine…&utm_content=note&utm_medium=seo_notes&utm_source=recommendation
# 安装注释包,固定格式
options()$repos
options()$BioC_mirror
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
# 引号中填写对应的注释包名,记得加“.db”
BiocManager::install("hugene10sttranscriptcluster.db",ask = F,update = F)
options()$repos
options()$BioC_mirror
9. 下载数据集GSE42872的表达矩阵,并且分别挑选出所有样本的(平均表达量/sd/mad/)最大的探针,并且找到它们对应的基因。
rm(list = ls())
options(stringsAsFactors = F)
# 加载并保存数据
f='GSE42872_eSet.Rdata'
library(GEOquery)
options( 'download.file.method.GEOquery' = 'libcurl' )
if(!file.exists(f)){
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F,
getGPL = F)
save(gset,file=f)
}
load('GSE42872_eSet.Rdata')
class(gset)
length(gset)
class(gset[[1]])
a=gset[[1]]
dat=exprs(a)
dim(dat)
pd=pData(a)
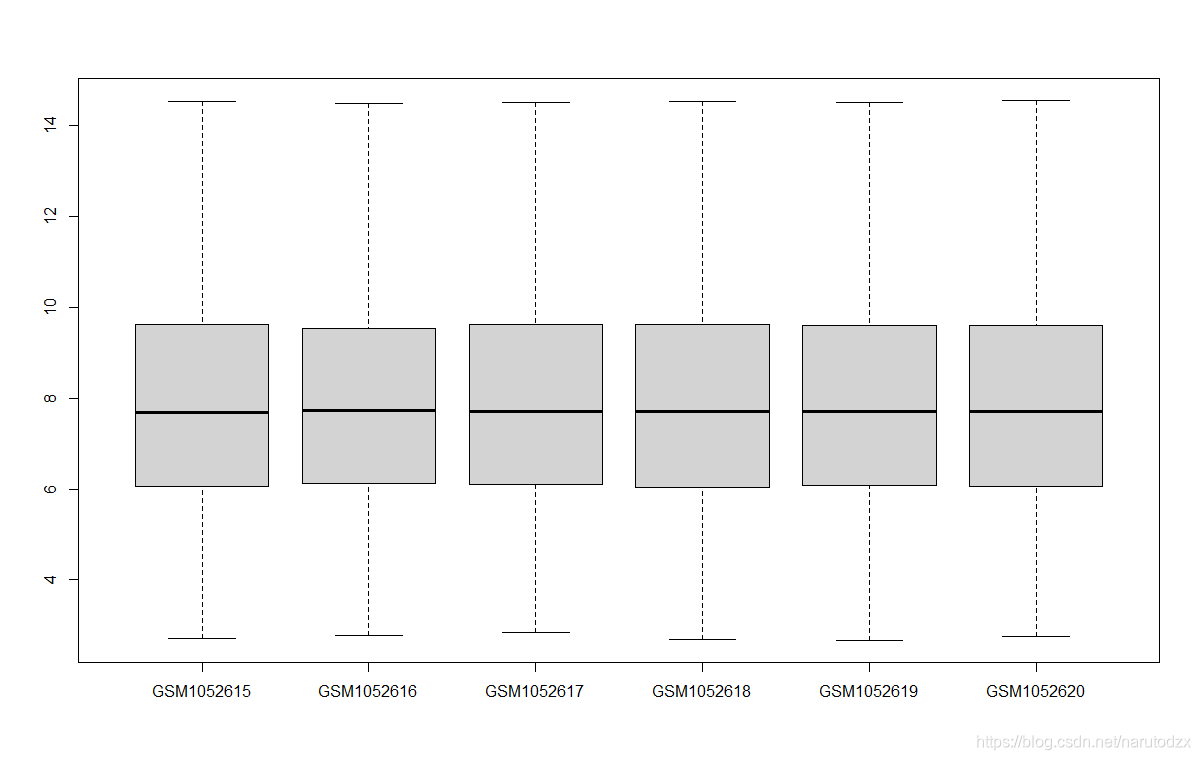
# 根据箱图可以大致看出数据的分布情况
boxplot(dat)
# 对数据的每行取mean,sd,mad,然后按降序排列,再取第一个最大值
sort(apply(dat,1,mean),decreasing = T)[1]
sort(apply(dat,1,sd),decreasing = T)[1]
sort(apply(dat,1,mad),decreasing = T)[1]

10. 下载数据集GSE42872的表达矩阵,并且根据分组使用limma做差异分析,得到差异结果矩阵。
rm(list = ls())
options(stringsAsFactors = F)
# 同第9题
f='GSE42872_eSet.Rdata'
library(GEOquery)
if(!file.exists(f)){
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file=f)
}
load('GSE42872_eSet.Rdata')
class(gset)
length(gset)
class(gset[[1]])
a=gset[[1]]
dat=exprs(a)
dim(dat)
pd=pData(a)
boxplot(dat)
# 设置分组信息
group_list=unlist(lapply(pd$title,function(x){
strsplit(x,' ')[[1]][4]
}))
group_list
# 设计比较矩阵
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(dat)
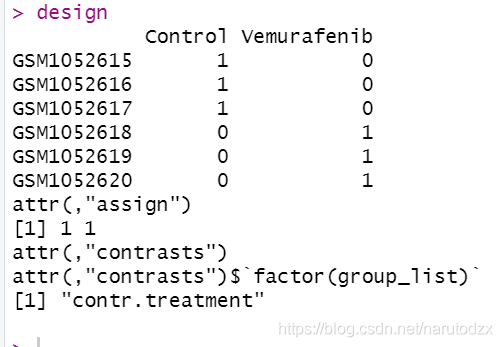
design
# 自定义比较元素
contrast.matrix<-makeContrasts(paste0(unique(group_list),collapse = "-"),levels = design)
contrast.matrix
# 参考:https://www.jianshu.com/p/d450f35db1cd
# 没看懂......
# step1
fit <- lmFit(dat,design)
# step2
fit2 <- contrasts.fit(fit, contrast.matrix) ##这一步很重要,大家可以自行看看效果
fit2 <- eBayes(fit2) ## default no trend !!!
##eBayes() with trend=TRUE
# step3
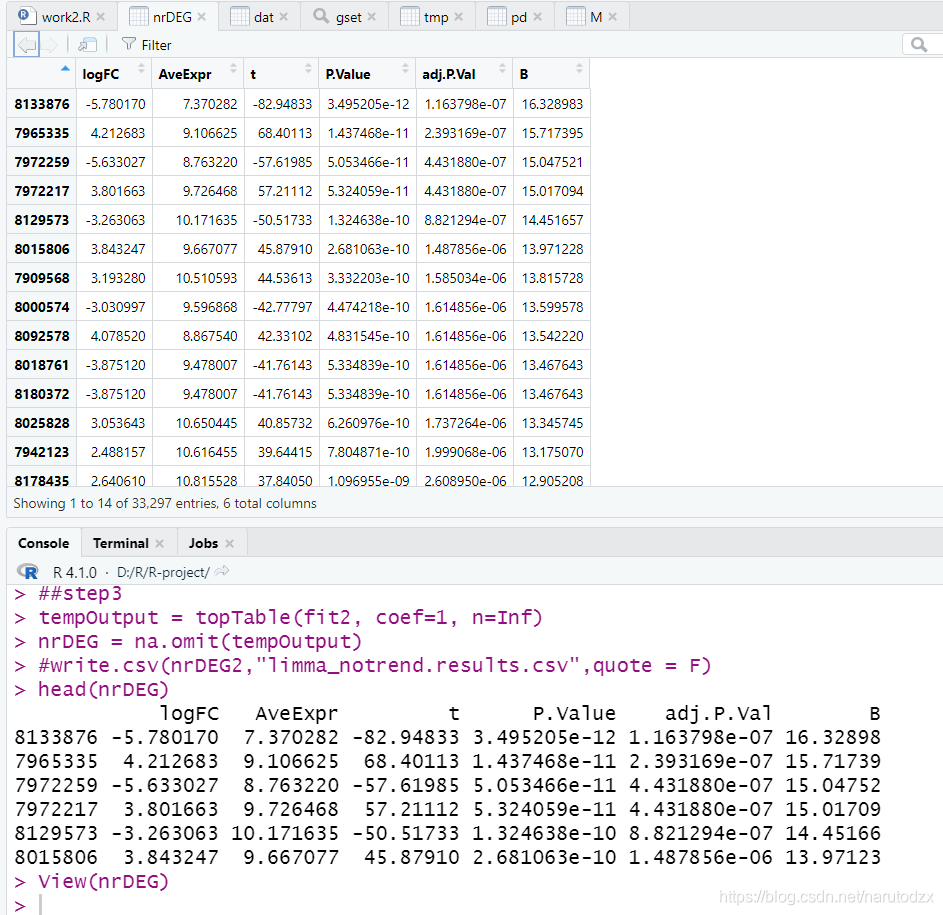
tempOutput = topTable(fit2, coef=1, n=Inf)
nrDEG = na.omit(tempOutput)
#write.csv(nrDEG2,"limma_notrend.results.csv",quote = F)
head(nrDEG)