初探芯片表达谱分析
实验目的
- 熟悉表达谱数据库的查询和数据下载
- 熟悉芯片表达谱数据分析的一般流程
- 掌握表达差异分析和基因富集分析的方法
- 了解常用的数据可视化方法
实验内容
-
GEO数据库查询和数据下载
- 使用R包
limma进行差异表达分析
- 使用R包
clusterProfiler进行基因富集分析
- 使用
gplots、ggpubr、pheatmap等R包对差异表达和富集分析进行结果可视化
实验题目
以GSE46456为例,该实验使用的芯片平台为 GPL198,拟南芥样本基因型包括:野生型、BRI1 单突变型、GUL2单突变型、BRI和 GUL 双突变型,每种基因型设置三种重复。研究三种突变型样本与WT野生型样本哪些基因存在显著的差异表达。根据所提供的演示代码和相关文件,请完成以下任务:
- 对获得的芯片数据进行数据标准化、探针过滤、limma差异分析,写明每一步骤的代码、目的以及中间结果。
- 运用limma获得突变体和野生型的差异表达基因集,并阐述差异分析结果的各列含义。
- 对所有基因做GSEA富集分析;并对三组上调的差异表达基因(bri1-WT、gul2-WT、bri1_gul2-WT)做GO富集分析,并解释富集结果,如有图片请注明图注信息。
实验过程
数据的获取、解压与读取
首先我们要从GEO数据库中下载登录号为GSE46456的数据,直接搜索登录号就可以得到下面的页面,滑到最下面就可以直接下载了。

图1 在GEO数据库中进行查询
图2 在GEO数据库中下载数据
我们也可以直接用r语言GEOquery包的getGEOSuppFiles()下载。
workdir <- "D:/test"
setwd(workdir)
library(GEOquery)
# 下载文件
getGEOSuppFiles("GSE46456",baseDir = workdir)
这个数据使用的芯片平台为 GPL198,拟南芥样本基因型包括:野生型、BRI1 单突变型、GUL2单突变型、BRI和 GUL 双突变型,每种基因型设置三种重复。从这些文件的命名就可以直观看到基因型。
图3 数据组成
下载并解压数据后,我们用Affy包的ReadAffy()读取CEL文件,将其处理成AffyBatch对象。
CEL文件是由Affymetrix DNA微阵列图像分析软件创建的数据文件。它包含从Affymetrix基因芯片上的“探针”中提取的数据,可以存储数千个数据点,这可能使其文件大小较大。CEL文件可以通过软件算法进行处理,并在二维网格上可视化,作为整个基因组实验的一部分。
[CEL File Extension - What is a .cel file and how do I open it? (fileinfo.com)](https://fileinfo.com/extension/cel#:~:text=A CEL file is a,it large in file size.)
数据预处理(背景纠正、标准化和探针信号汇总等)
芯片数据的预处理流程主要可以分为四个步骤:
- background correction:移除非特异结合等背景噪音,有助于检出较低丰度下的倍数差异。
- normalization:消除测量间的非实验误差,使得实验条件下的测量可以相互比较。
- pm correction(部分方法无此步):对pm探针的荧光值进行修正。
- summarization:将前面得到的荧光强度值从探针水平汇总到探针组水平
三种常见预处理的方法:
- 使用affy包 EXPRESSO函数。
- 使用affyPLM包 threestep 函数(返回值经过log2转换)
- 使用一体化函数,如:rma (affy包)、gcrma (gcrma包)、mas5 (affy包)等。
三种方法的特性:
- 前两种方法可以用于自由组合不同的处理方法对数据进行标准化。但一般不建议这么做,因为许多处理方法不能整合到一起,建议使用一体化标准方法rma()等。
- 一体化函数中,mas5返回结果没有log2转换,rma和gcrma结果经过log2转换,而后续大多数分析都是依赖log2转换的结果;gcrma算法使用了MM探针的信息,因此不适用于没有MM探针的部分affy芯片,适用范围窄。
- mas5是对单张芯片进行的标准化处理,rma是对多张芯片同时进行标准化处理。
我们使用第三种方法:rma (affy包)。这个函数的原理在这篇论文里有详细说明。

图4 rma函数的原论文
comparison of normalization methods for high density oligonucleotide array data based on variance and bias | Bioinformatics | Oxford Academic (oup.com)
Bolstad, B M et al. “A comparison of normalization methods for high density oligonucleotide array data based on variance and bias.” Bioinformatics (Oxford, England) vol. 19,2 (2003): 185-93. doi:10.1093/bioinformatics/19.2.185
# 生成文件列表,以便批量导入文件
cels <- list.files(paste(workdir,"/GSE46456/GSE46456_RAW",sep = ''),pattern="*.gz",full.names =TRUE)
celfiles <- ReadAffy(filenames=cels)# 读取CEL文件,将其处理成AffyBatch对象
celfiles.rma <- rma(celfiles)# 将AffyBatch对象转换为ExpressionSet对象,对数据进行标准化
# ?rma
cols <- brewer.pal(8,"Set1")
par(mfrow=c(2,2))
boxplot(celfiles, col=cols)
boxplot(celfiles.rma, col=cols)
hist(celfiles, col=cols)#密度和对数强度直方图
hist(celfiles.rma, col=cols)
处理前后数据质量比较如下图所示,可以看到处理之后,数据的分布更加趋同了。

图5 处理前后数据分布对比
数据过滤(探针过滤)
表达谱芯片上的探针,理论上能够覆盖到所有基因,一个细胞中不可能所有基因都同时表达,仅有少数基因同时表达,探针与目标之间存在着非特异性结合,如果不过滤低表达探针会造成后续分析结果的假阳性。
探针过滤的方法:
- genefilter包中的nsFilter函数提供了一站式的探针过滤,能够一步对ExpressionSet的探针进行过滤,返回一个list。list中的eset为过滤后的ExpressionSet,filter.log为每一步过滤到多少探针的记录。
- mas5calls 函数整合在 affy 包中,这个函数的功能是对探针状态进行评估,分类为“P”,“M”和“A”。该方法可以只提取其中分类为“P”的探针,认为其他探针不表达;亦或是只删去分类为“A”的探针。由于 mas5calls 函数只能针对 AffyBatch 对象,因此只能用于标准化前的数据。
我们使用第一种方法进行探针过滤。
图6 nsFilter函数官方文档
我们使用默认参数,也就是过滤掉没有ENTREZ ID的探针,如果有多个探针对应到同一ENTREZ ID的时候,留下var.func值最大的探针,其余过滤。截断值设置为0.5,也就是说过滤到50%的基因。
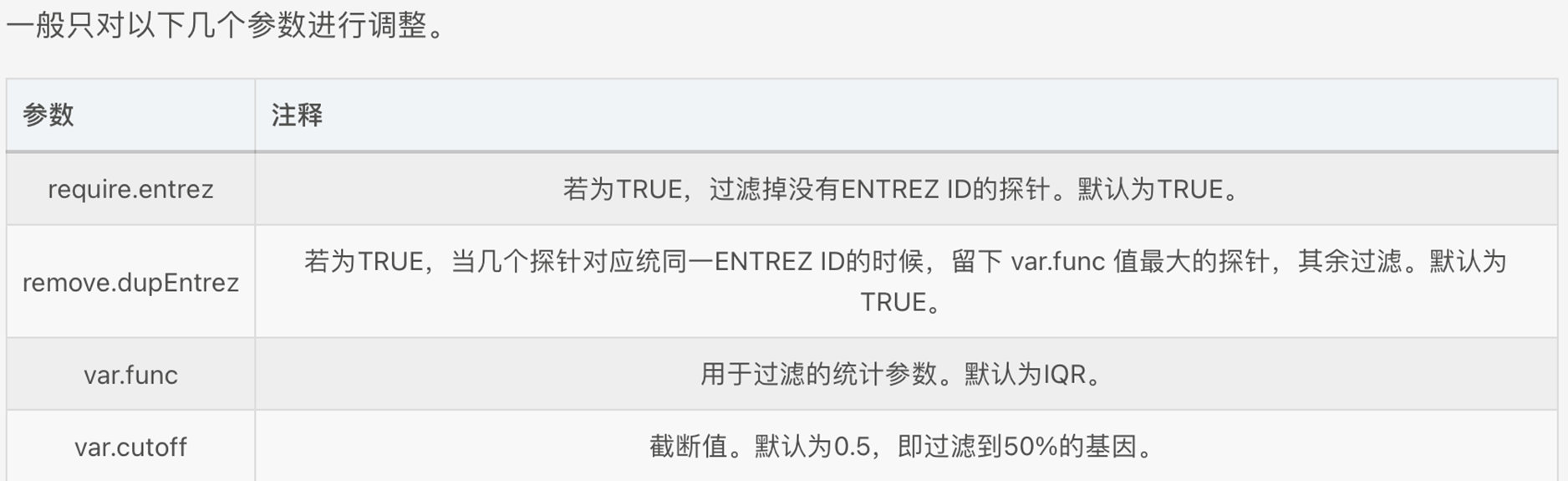
图7 nsFilter函数参数注释
# list中的eset为过滤后的ExpressionSet,filter.log为每一步过滤到多少探针的记录
celfiles.filtered <- nsFilter(celfiles.rma, require.entrez=FALSE, remove.dupEntrez=FALSE)
celfiles.filtered$filter.log
celfiles.filtered$eset
# 获得表达量矩阵
eset <- exprs(celfiles.filtered$eset)
下面是表达量矩阵,每一行是一个探针(基因),每一列是一个样本(基因型)。
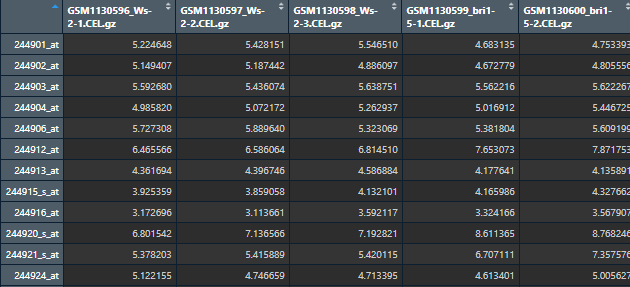
图8 表达量矩阵
探针注释(添加基因注释信息)
芯片厂商不会使用基因名作为探针的名称,而是使用自己定义的探针名称。要合并重复探针,我们必须先对探针进行注释,确定每个探针对应检测哪个基因的表达,然后再合并重复探针。而后续分析如GSEA,只能对基因进行分析,因此也要求对探针进行注释。
我们用这个探针的注释信息进行对应的连接操作即可。

图9 探针的注释信息
ara_anno <- read.delim("affy_ATH1_array_elements-2010-12-20.txt")
ids <- match(rownames(eset), ara_anno$array_element_name)
rownames(eset) <- ara_anno$locus[ids]
colnames(eset) <- sub(".CEL.gz","",colnames(eset))
下面是注释后的表达量矩阵,每一行是基因,每一列是样本。
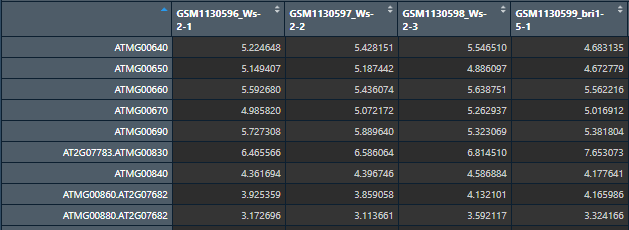
图10 注释后的表达量矩阵
limma差异分析

图11 limma差异分析的步骤
首先我们构造分组矩阵,总共有四个基因型,每组重复三次。
group_list <- c(rep('wt',3),rep('bri1',3),rep('gul2',3),rep('gul_bri',3))
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(eset)
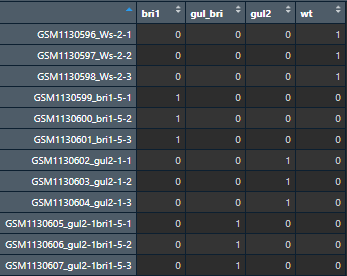
图12 limma差异分析的分组矩阵
接下来构建对照矩阵,也就是和野生型作比较,共有3组,这里的顺序很重要,野生型需要放到后面。
contrast.matrix<- makeContrasts(bri1-wt,gul2-wt,gul_bri-wt,levels =design)
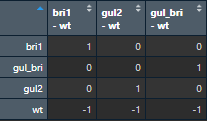
图13 limma差异分析的对照矩阵
接下来就进入了limma“三部曲”,也就是拟合、差值计算和检验。
#limma"三部曲"
fit1 <- lmFit(eset,design)#线性模型拟合
fit2 <- contrasts.fit(fit1,contrast.matrix)#根据对比模型进行差值计算
fit2 <- eBayes(fit2)#贝叶斯检验
接下来我们输出差异表达基因。
#利用toptable 导出DEG结果
limma_results <- lapply(colnames(contrast.matrix),function(x){topTable(fit2,coef=x,adjust="fdr",sort.by="logFC",number=Inf)})
names(limma_results) <- colnames(contrast.matrix) #对导出的结果标记title信息
save(limma_results,file="limma_compare_res.RData")
#对每对比较的样本对DEG结果单独导出DEG信息6
for (n in names(limma_results)){
write.table(limma_results[[n]],file = sprintf('%s.tsv',gsub(' ','',n)),row.names =FALSE,sep="\t")
}
save(eset,file='eset.RData')
总共有三组结果,我们随便取一组来看看。总共有7列,每一列的含义如下:
-
ID: Gene ID
-
logFC: 两组表达值之间以2为底对数化的变化倍数(Fold change, FC),由于基因表达矩阵本身已经取了对数,这里实际上只是两组基因表达值均值之差
-
AveExpr:该探针组所在所有样品中的平均表达值
-
t:贝叶斯调整后的两组表达值间 T 检验中的 t 统计量
-
P.Value: 检验P值
-
adj.P.Val:调整后的 P 值(多重检验BH等方法)
-
B:是经验贝叶斯得到的标准差的对数化值
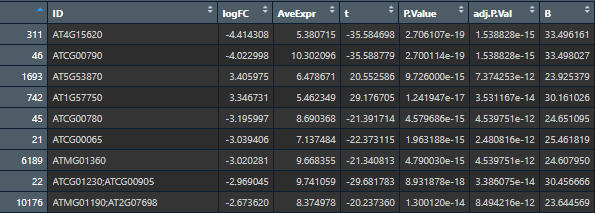
图14 差异表达基因的信息
差异表达基因结果的可视化
我们通过热图和火山图对差异表达分析的结果进行可视化。从下面的热图可以看到,其实差异肉眼看上去并不是很显著。火山图共有六组。
# 换个名字,实在太长了
for (i in 1:ncol(eset)){
colnames(eset)[i] <- strsplit(colnames(eset)[i],'_')[[1]][2]
}
png(filename = 'heatmapDEG.png',width = 3580,height = 2200)
pheatmap(eset, col=c(colorRampPalette(brewer.pal(9,"Blues")[7:2])(100),colorRampPalette(brewer.pal(9,"Reds")[2:7])(100)),border_color=NA,cluster_rows=T,cluster_cols=T,show_rownames=F,show_colnames=T,angle_col=315,fontsize=13,main="expression",display_numbers=F)
dev.off()
# 火山图
library(ggpubr)
library(ggthemes)
for (n in names(limma_results)){
file_name <- sprintf('%s.tsv',gsub(' ','',n))
deg.data <- read.table(file_name,header = T,sep = "\t")
deg.data$logP <- -log10(deg.data$adj.P.Val) #-log10值转换
#head(deg.data)
deg.data$Group = "not-significant" #定义Group列
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC > 2))] = "up-regulated" #定义DEG标准
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC < -2))] = "down-regulated" #定义DEG标准
table(deg.data$Group) #统计DEG数量
t <- ggscatter(deg.data,x="logFC",y = "logP",color = "Group")+theme_base()
ggscatter(deg.data,x="logFC",y = "logP",color = "Group")
#新加一列lable
deg.data$Lable = ""
#对差异表达基因P值从小到大排序
deg.data <- deg.data[order(deg.data$adj.P.Val),]
#从高表达基因中选取adj.P.Val最显著的10个基因
up.genes <- head(deg.data$ID[which(deg.data$Group == "up-regulated")],10)
#从低表达基因中选取adj.P.Val最显著的10个基因
down.genes <- head(deg.data$ID[which(deg.data$Group == "down-regulated")],10)
#讲上两步选取的显著基因合并并加入到lable中
deg.top10.genes <- c(as.character(up.genes),as.character(down.genes))
deg.data$Lable[match(deg.top10.genes,deg.data$ID)] <- deg.top10.genes
png(filename = paste(file_name,"deg_volcano.png",sep = ''),width = 3580,height = 2200,res = 300)
deg_volcano <- ggscatter(deg.data,x="logFC",y = "logP",color = "Group"
,palette = c("#2f5688","#BBBBBB","#CC0000")
,size = 1,label = deg.data$Lable,font.label = 8
,repel = T,xlab = "Log2FoldChange"
,ylab="-Log10(Adjust P-value)",) +
theme_base()+geom_hline(yintercept = 1.30, linetype="dashed") +
geom_vline(xintercept = c(-2,2),linetype="dashed") +
ggtitle(n)
print(deg_volcano)
dev.off()
}
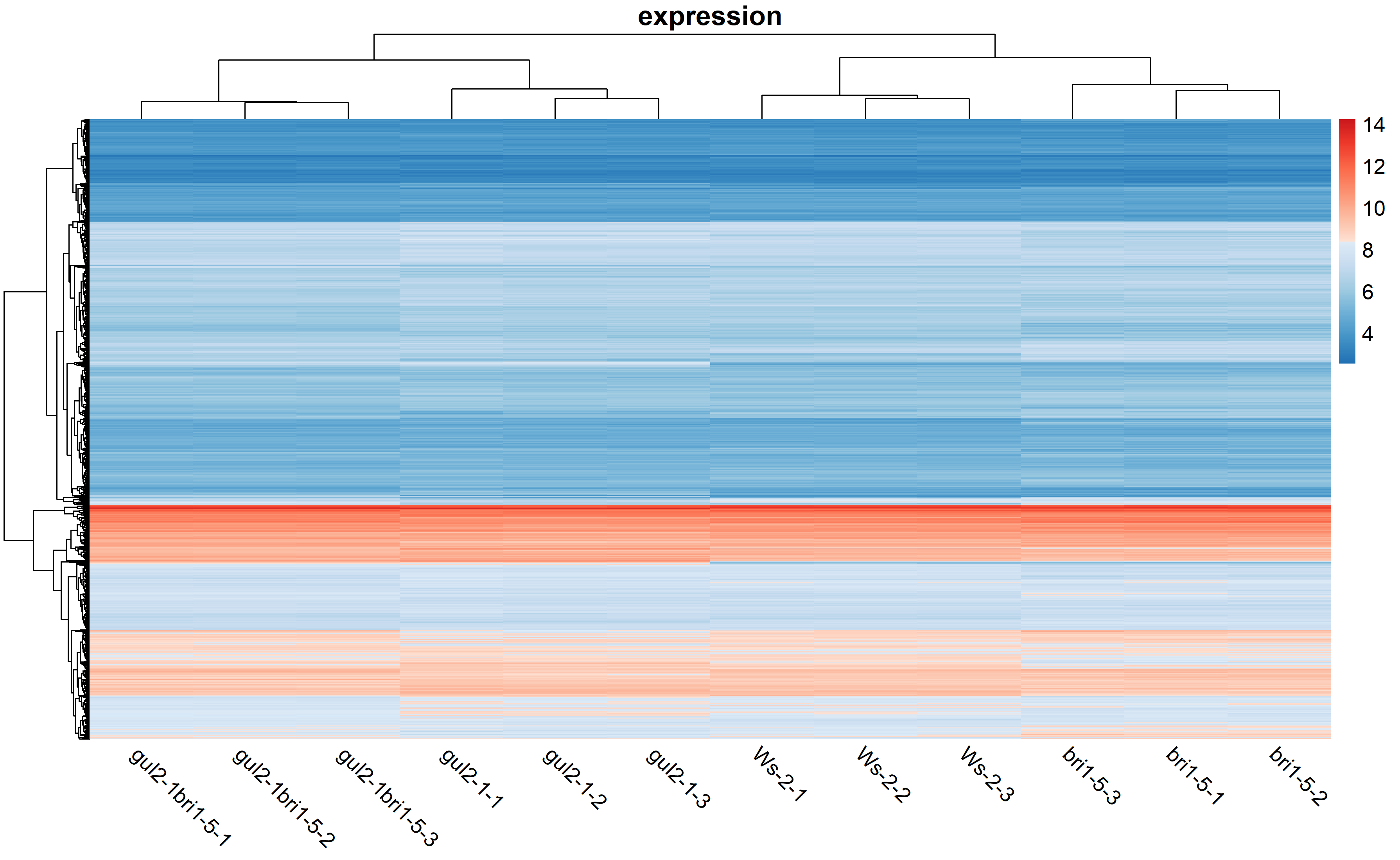
图15 差异表达热图
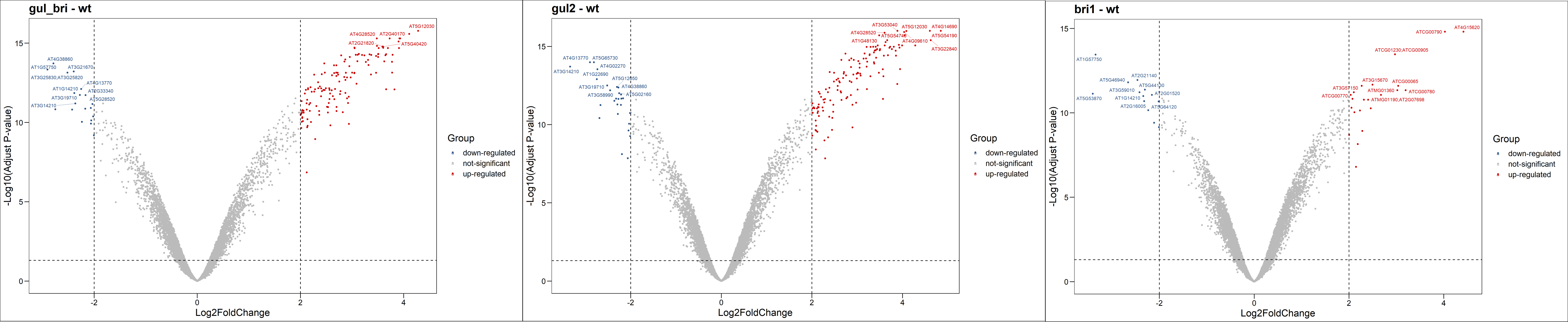
图16 差异表达火山图
富集分析及其结果可视化
首先我们进行GSEA,除了基础的图外,我们还将富集结果的前四名拿出来单独画图。下面的表是富集结果。
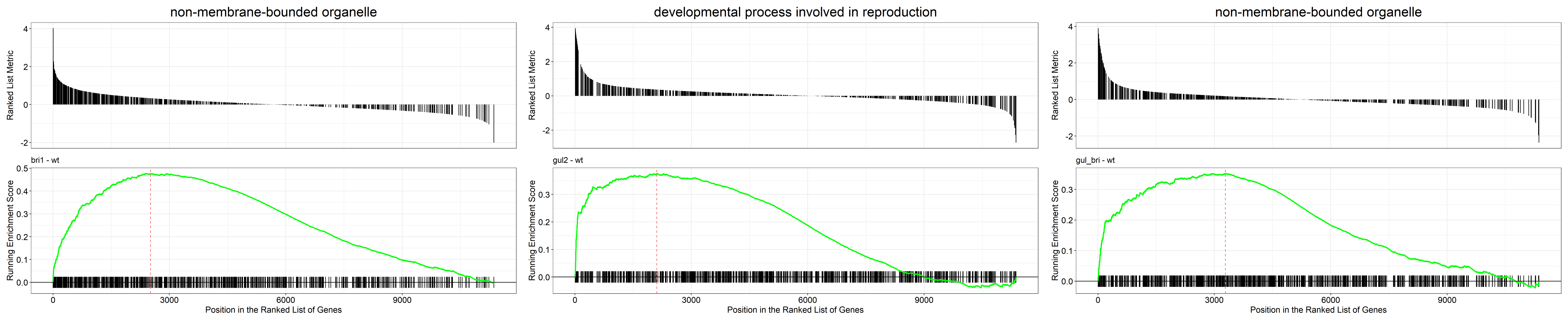
图17 GSEA(GO)结果图1
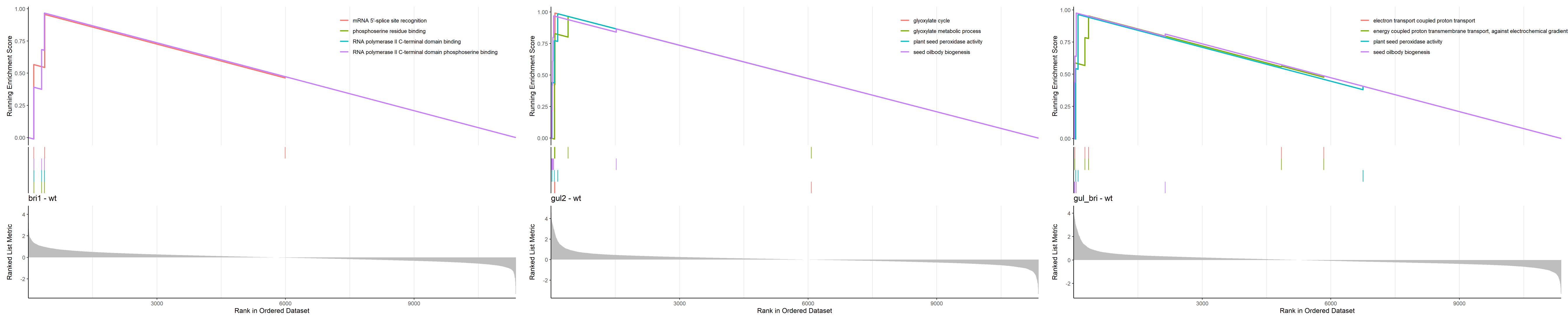
图17 GSEA(GO)结果图2
表1 GSEA(GO)结果
| ONTOLOGY | ID | Description | setSize | enrichmentScore | NES | pvalue | p.adjust | qvalues | rank | leading_edge | core_enrichment | | -------- | ---------- | ------------------------------------------------------------ | ------- | --------------- | ----------- | ----------- | ----------- | ----------- | ---- | ------------------------------ | ------------------------------------------------------------ | | BP | GO:0010344 | seed oilbody biogenesis | 6 | 0.976030854 | 1.98297256 | 0.00020284 | 0.00020284 | 0.014726679 | 64 | tags=83%, list=1%, signal=83% | AT1G05510/AT5G40420/AT4G25140/AT3G27660/AT3G01570 | | MF | GO:1990137 | plant seed peroxidase activity | 3 | 0.964308935 | 1.651721829 | 0.002232596 | 0.002232596 | 0.046555276 | 104 | tags=67%, list=1%, signal=66% | AT4G26740/AT5G55240 | | BP | GO:0015988 | energy coupled proton transmembrane transport, against electrochemical gradient | 5 | 0.953725747 | 1.850294415 | 0.000204332 | 0.000204332 | 0.014726679 | 347 | tags=60%, list=3%, signal=58% | ATMG01360/ATCG00430/ATMG00580 | | BP | GO:0015990 | electron transport coupled proton transport | 5 | 0.953725747 | 1.850294415 | 0.000204332 | 0.000204332 | 0.014726679 | 347 | tags=60%, list=3%, signal=58% | ATMG01360/ATCG00430/ATMG00580 | | BP | GO:0009647 | skotomorphogenesis | 3 | 0.948351246 | 1.624388612 | 0.006494824 | 0.006494824 | 0.098872054 | 122 | tags=67%, list=1%, signal=66% | AT5G54190/AT2G46990 | | MF | GO:1990404 | protein ADP-ribosylase activity | 3 | 0.936487083 | 1.604067017 | 0.009742237 | 0.009742237 | 0.133204734 | 83 | tags=33%, list=1%, signal=33% | AT5G22470 | | BP | GO:0042149 | cellular response to glucose starvation | 3 | 0.936059807 | 1.603335154 | 0.009742237 | 0.009742237 | 0.133204734 | 730 | tags=100%, list=6%, signal=94% | AT1G15330/AT1G80090/AT4G27460 | | BP | GO:0019915 | lipid storage | 16 | 0.926634447 | 2.398720888 | 0.000189825 | 0.000189825 | 0.014726679 | 268 | tags=62%, list=2%, signal=61% | AT1G05510/AT5G40420/AT4G25140/AT2G25890/AT4G26740/AT3G27660/AT3G01570/AT5G51210/AT3G18570/AT5G56100 |
然后我们再对对三组上调的差异表达基因(bri1-WT、gul2-WT、bri1_gul2-WT)做GO富集分析。
BRI1 单突变型在核糖体上富集,可能该突变的上调基因均编码核糖体的相关元件。GUL2单突变型在抗寒、抗旱方面富集,很有可能获得了抗逆性增强的基因型。双突变则在抗寒和抗低氧方面富集,也有可能具有良好的抗逆性。
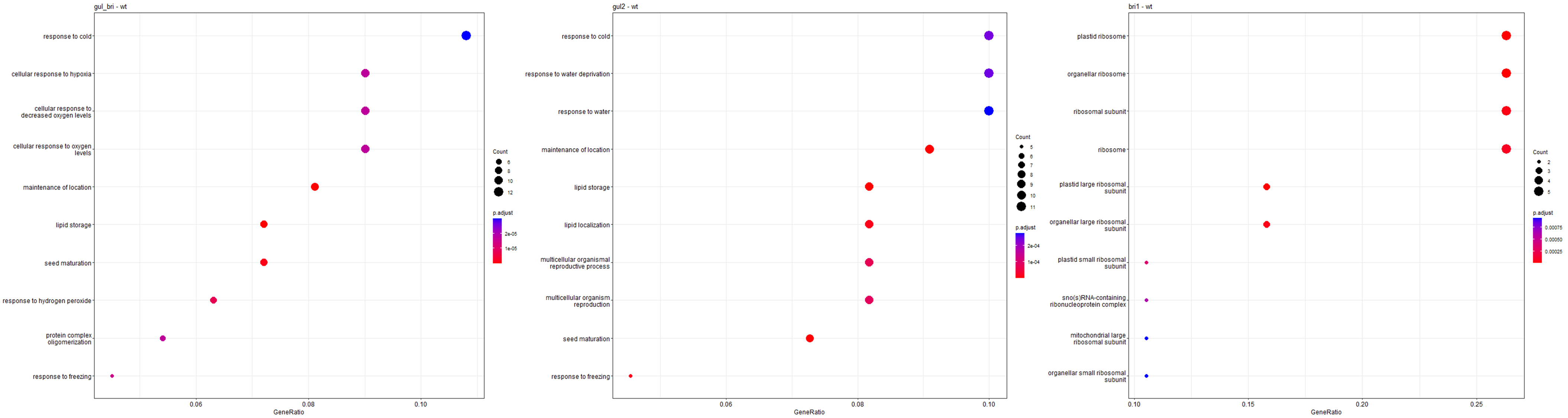
图18 GO富集分析结果图
for (n in names(limma_results)){
file_name <- sprintf('%s.tsv',gsub(' ','',n))
GSEA_data <- read.table(file_name,header = T,sep = "\t")
GSEA_gene_lists <- GSEA_data$logFC #提取表达量变化值
names(GSEA_gene_lists) <- GSEA_data$ID #给提取出来的值赋予ID
GSEA_gene_lists <- sort(GSEA_gene_lists, decreasing = TRUE)#降序排列
organisms <- get("org.At.tair.db") #获取拟南芥数据库信息
GSEA_GO_Result <-
gseGO(
geneList = GSEA_gene_lists,
ont = "ALL",
keyType = "TAIR",
nPerm = 10000,
minGSSize = 3,
maxGSSize = 800,
pvalueCutoff = 0.05,
verbose = TRUE,
OrgDb = organisms ,
pAdjustMethod = "none"
)
Sort_GO_result <- GSEA_GO_Result[order(GSEA_GO_Result$enrichmentScore, decreasing = T),]
write.table(Sort_GO_result,paste(file_name,'GSEA_GO_Result.txt',sep = ''),sep = "\t",quote = F,col.names = T,row.names = F)
GSEA_GO_Result_plot <- gseaplot2(GSEA_GO_Result, row.names(Sort_GO_result)[1:4])+ggtitle(n)
png(filename = paste(file_name,'GSEA_GO_Result.png',sep = ''),width = 3580,height = 2200,res = 300)
print(GSEA_GO_Result_plot)
dev.off()
GSEA_GO_Result_plot2 <- gseaplot(GSEA_GO_Result,by="all",title = GSEA_GO_Result$Description[1],geneSetID=1)+ggtitle(n)
png(filename = paste(file_name,'GSEA_GO_Result2.png',sep = ''),width = 3580,height = 2200,res = 300)
print(GSEA_GO_Result_plot2)
dev.off()
}
for (n in names(limma_results)){
file_name <- sprintf('%s.tsv',gsub(' ','',n))
deg.data <- read.table(file_name,header = T,sep = "\t")
deg.data$logP <- -log10(deg.data$adj.P.Val) #-log10值转换
deg.data$Group = "not-significant" #定义Group列
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC > 2))] = "up-regulated" #定义DEG标准
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC < -2))] = "down-regulated" #定义DEG标准
data<- deg.data[deg.data$Group=="up-regulated",]
ego <- enrichGO(gene = data$ID,keyType = "TAIR",OrgDb=organisms,ont="ALL",pAdjustMethod="BH",qvalueCutoff=0.05)
png(file = paste(file_name,'GO_dot.png',sep = ''),bg="transparent",width=1000,height=800)
GO_dot <- dotplot(ego,showCategory=10)+ggtitle(n)
print(GO_dot)
dev.off()
}
讨论
1. 为什么要对探针信号或基因表达量取对数?为什么是log2不是log10或ln?
原始的表达量矩阵离散程度很高。有的基因表达丰度比较高,counts数为10000,有些低表达的基因counts可能10,甚至在有些样本中为0。 即使经过了RPKM/FPKM等方法抵消了一些测序技术误差的影响,但高低丰度基因的表达量的差距依然很大。
我们关注的是数据的相对关系,而对数函数在定义域是单调递增,取对数之后不会改变对数的相对关系。取对数之后可以缩小数据的绝对数值,方便计算。取对数之后压缩了变量的极差,数据更加平稳,削弱了模型的共线性、异方差性。
至于为什么用log2,是因为我们认为基因表达量增加1倍就可以造成生物学上的一些变化,比如在生物学上会有一些和“二”或者“一半”相关的概念,半衰期还有S型曲线的最大增速点也是在一半。而且如果前面是对表达量取log2,后面计算fold-chage就可以直接相减。
2. 为什么要设置生物学重复?
生物重复是指对同一个处理组中独立来源的多个样本分别进行独立测定分析,是整个实验的完全重复,如将具有同一基因型的多个细胞株进行独立地测定。由于遗传和环境等因素的影响会引起有机体的个体差异,因此需要采用生物重复的实验设计方法来消除该差异。
设计生物重复可以:
- 能够消除组内误差:生物学重复可以测量变异程度;
- 增强结果的可靠性:测序的样本数越多,越能够降低背景差异;
- 检测离群样本:异常样本的存在,会严重影响测序结果的准确性,通过计算样本间的相关性可以发现异常样本,将其排除。
ased on variance and bias | Bioinformatics | Oxford Academic (oup.com)](https://academic.oup.com/bioinformatics/article/19/2/185/372664)
Bolstad, B M et al. “A comparison of normalization methods for high density oligonucleotide array data based on variance and bias.” Bioinformatics (Oxford, England) vol. 19,2 (2003): 185-93. doi:10.1093/bioinformatics/19.2.185