今天开始学习使用FastQC软件对范例SRA测序文件的质量进行分析。
主要参考文章:
RNA-seq(3):sra到fastq格式转换并进行质量控制
转录组入门(3):了解fastq测序数据
用FastQC检查二代测序原始数据的质量
FastQC Tutorial & FAQ
从零开始完整学习全基因组测序(WGS)数据分析:第2节 FASTA和FASTQ
数据质控是一个综合的评价标准,其中主要指标为碱基质量与含量分布,如果这两个指标合格,后面大部分指标都可以通过;如果这两项不合格,其余都会受到影响。
其中一些指标并不适合所有数据,例如DNA测序数据与RNA测序数据之间的差异等,要根据具体数据类型具体分析。
FASTA的介绍
我们接触到的序列信息有FASTA和FASTQ两种格式,这是存储核苷酸序列信息(DNA序列)或者蛋白质序列信息最常使用的两种纯文本文件。
FASTA存的都是已经排列好的序列(如参考序列),起源于一款“FASTA”的比对软件,之后便以FASTA作为这种存储有顺序的序列数据的文件后缀,文件后缀除了.fasta之外,也常用.fa或者.fa.gz(gz压缩),包括常用的参考基因组序列、蛋白质序列、编码DNA序列(coding DNA sequence,简称CDS)、转录本序列等文件。
FASTA文件主要由两个部分构成:序列头信息(有时包括一些其它的描述信息)和具体的序列数据。序列头信息独占一行,以大于号(>)开头作为识别标记,其中除了记录该条序列的名字之外,有时候还会接上其它的信息。紧接的下一行是具体的序列内容,直到另一行碰到另一个大于号(>)开头的新序列或者文件末尾。
>gene_00284728 length=231;type=dna
GAGAACTGATTCTGTTACCGCAGGGCATTCGGATGTGCTAAGGTAGTAATCCATTATAAGTAACATG
CGCGGAATATCCGGGAGGTCATAGTCGTAATGCATAATTATTCCCTCCCTCAGAAGGACTCCCTTGC
GAGACGCCAATACCAAAGACTTTCGTAAGCTGGAACGATTGGACGGCCCAACCGGGGGGAGTCGGCT
ATACGTCTGATTGCTACGCCTGGACTTCTCTT
FASTQ的介绍
FASTQ存的则是产生自测序仪的原始测序数据,它由测序的图像数据转换过来,也是文本文件,文件大小依照不同的测序量(或测序深度)而有很大差异,小的可能只有几M,大的则常常有几十G上百G,文件后缀通常都是.fastq,.fq或者.fq.gz(gz压缩)。
FASTQ有独特的格式:每四行成为一个独立的单元,我们称之为read。具体的格式描述如下:
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是 每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
@DJB775P1:248:D0MDGACXX:7:1202:12362:49613
TGCTTACTCTGCGTTGATACCACTGCTTAGATCGGAAGAGCACACGTCTGAA
+
JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD?DDDDDDBDDDABDDCA
@DJB775P1:248:D0MDGACXX:7:1202:12782:49716
CTCTGCGTTGATACCACTGCTTACTCTGCGTTGATACCACTGCTTAGATCGG
+
IIIIIIIIIIIIIIIHHHHHHFFFFFFEECCCCBCECCCCCCCCCCCCCCCC
FastQC的介绍
FastQC aims to provide a simple way to do some quality control checks on raw sequence data coming from high throughput sequencing pipelines. It provides a modular set of analyses which you can use to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis.
The main functions of FastQC are:
- Import of data from BAM, SAM or FastQ files (any variant)
- Providing a quick overview to tell you in which areas there may be
problems - Summary graphs and tables to quickly assess your data
- Export of results to an HTML based permanent report
- Offline operation to allow automated generation of reports without running the interactive application
1. 首先进入待分析数据的目录中,查看需要分析的SRA名称:
xiaomotong@VirtualBox: cd ~/Public/SRA/sra
xiaomotong@VirtualBox:~/Public/SRA/sra$ ll
总用量 2262608
drwxrwxr-x 2 xiaomotong xiaomotong 4096 5月 17 15:04 ./
drwxrwxr-x 8 xiaomotong xiaomotong 4096 5月 14 15:40 ../
-rw-rw-r-- 1 xiaomotong xiaomotong 1057984832 5月 14 12:02 SRR390728_1.fastq
-rw-rw-r-- 1 xiaomotong xiaomotong 1057984832 5月 14 12:02 SRR390728_2.fastq
-rw-rw-r-- 1 xiaomotong xiaomotong 195044834 5月 14 11:40 SRR390728.sra
-rw-rw-r-- 1 xiaomotong xiaomotong 5867015 5月 14 11:40 SRR390728.sra.vdbcache
2. 使用fastqc命令对目标数据进行分析:
xiaomotong@VirtualBox:~/Public/SRA/sra$ fastqc -t 3 ./SRR390728_1.fastq
Started analysis of SRR390728_1.fastq
Approx 5% complete for SRR390728_1.fastq
。。。。。。
Approx 95% complete for SRR390728_1.fastq
Analysis complete for SRR390728_1.fastq
fastqc 命令的用法
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
#参数:
-o --outdir 输出目录,需自己创建目录
–(no)extract 是否解压输出文件,默认是自动解压缩zip文件。加上–noextract不解压文件。
-f 指定输入文件的类型,支持fastq|bam|sam三种格式的文件,默认自动识别。
-t --threads选择程序运行的线程数,即同时处理的文件数目。
-c --contaminants,污染物选项,输入的是一个文件,格式是Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到。
fastqc -o . *.fastq.gz
3. 质控结果解读
处理后产生.html和fastqc.zip两个文件,如下:
xiaomotong@VirtualBox:~/Public/SRA/sra$ ll
总用量 2264264
drwxrwxr-x 2 xiaomotong xiaomotong 4096 5月 19 15:21 ./
drwxrwxr-x 8 xiaomotong xiaomotong 4096 5月 14 15:40 ../
-rw-rw-r-- 1 xiaomotong xiaomotong 1057984832 5月 14 12:02 SRR390728_1.fastq
-rw-rw-r-- 1 xiaomotong xiaomotong 526256 5月 19 15:20 SRR390728_1_fastqc.html
-rw-rw-r-- 1 xiaomotong xiaomotong 316413 5月 19 15:20 SRR390728_1_fastqc.zip
报告保存在.html文件中,可以调用Linux系统上的firefox命令来打开(参考文章)
xiaomotong@VirtualBox:~/Public/SRA/sra$ firefox ./SRR390728_1_fastqc.html
3.1. FastQC报告概览
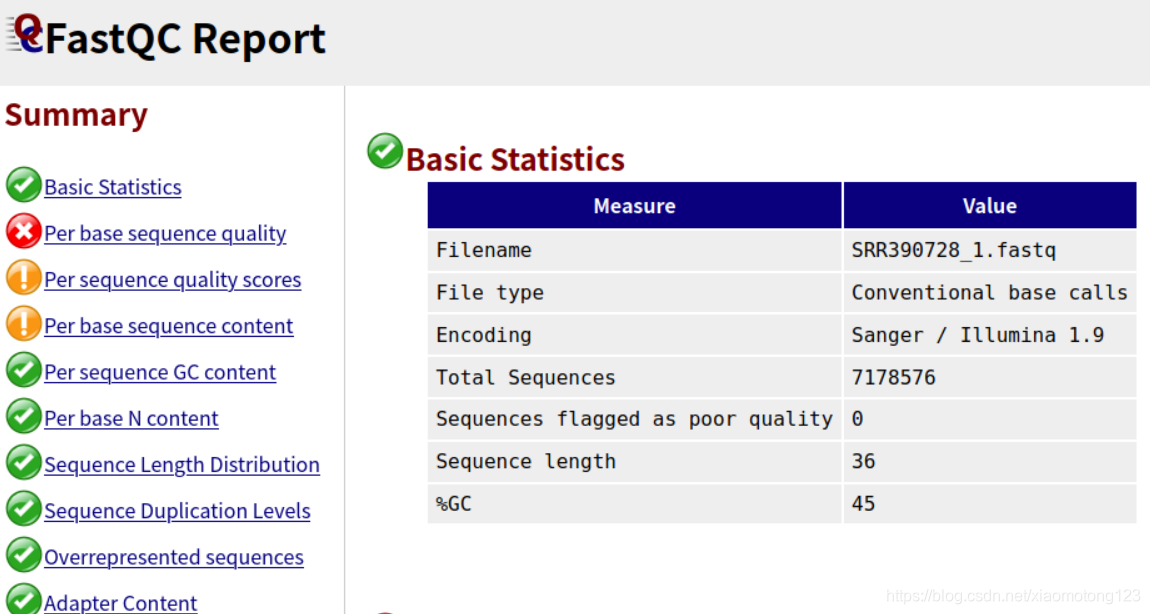
- 左边是目录概要,可以点击想要看的结果,右边会跳转到特定详细的可视化结果。绿色代表“通过”,黄色代表“警告”,红色代表“不通过,失败”。
- 右边是Basic Statistics,基本的数据统计包括文件名,文件类型,编码形式,总的序列数,质量差的序列,序列平均长度,GC含量。
3.2. 每个位置的碱基测序质量

- Per base sequence quality,每个read各位置碱基的测序质量。横轴是碱基的位置,纵轴是质量分数,Quality
score=-10log10p(p代表错误率),所以当质量分数为40的时候,p就是0.0001,质量算高了。红色线代表中位数,蓝色线代表平均数,黄色线是25%-75%区间,触须是10%-90%区间(黄色和触须我不是特别明白)。若任一位置的下四分位数低于10或者中位数低于25,出现“警告”;若任一位置的下四分位数低于5或者中位数低于20,出现“失败,Fail”。通常认为从第二个碱基开始,平均每个碱基的测序质量boxplot下四分位线在30分以上,则认为测序质量非常好。
3.3. 每条序列的测序质量分数

- Per sequence quality scores,reads质量的分布,当峰值小于27时,警告;当峰值小于20时,fail。
- 一般认为,90%的reads测序质量在35分以上,则认为该测序质量非常好。
3.4. ATGC碱基在各个位置上的分布

- Per base sequence content,对所有reads的每一个位置,统计ATCG四种碱基的分布,横轴为位置,纵轴为碱基含量,正常情况下每个位置每种碱基出现的概率是相近的,四条线应该平行且相近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。本结果前10个位置,每种碱基频率有略微的差别,说明可能有污染。当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
- 一般来说,AT含量高于CG含量,AT含量约28%,CG含量约22%。由于测序问题,通常第一二位置的碱基测序质量比较低,ATCG含量也不正常。这种情况不影响数据质量,如果实在介意,可在后续bowtie mapping的时候将前两个碱基去掉。
3.5. GC碱基在各个位置上的分布

- Per Sequence GC Content,统计reads的平均GC含量的分布。红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
3.6. N碱基(无法识别的碱基)在各个位置上的分布

- Per base N content,当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”,统计N的比率。正常情况下,N值非常小,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"。
3.7. reads长度分布

- Sequence Length Distribution,reads长度分布,当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”。
3.8. 序列不同拷贝数的水平

- Sequence Duplication Levels,统计不同拷贝数的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在。****横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。如果原始数据很大(事实往往如此),做这样的统计将非常慢,所以fastqc中用fq数据的前200,000条reads统计其在全部数据中的重复情况。如果重复数目大于等于10的reads被合并统计,我们会看到最右侧略有上扬。当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“。
3.9. 一条序列的重复数(此条选取参考文章中数据)
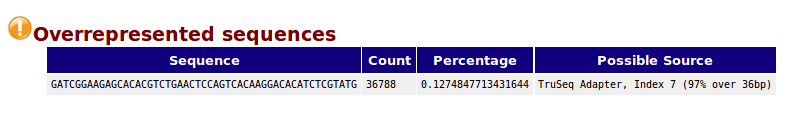
- Overrepresented sequences,一条序列的重复数,因为一个转录组中有非常多的转录本,一条序列再怎么多也不太会占整个转录组的一小部分(比如1%),如果出现这种情况,不是这种转录本巨量表达,就是样品被污染。这个模块列出来大于全部转录组1%的reads序列,但是因为用的是前200,000条,所以其实参考意义不大,完全可以忽略。
- 如果有某个序列大量出现,就叫做over-represented。fastqc的标准是占全部reads的0.1%以上。和上面的duplicate analysis一样,为了计算方便,只取了fq数据的前200,000条reads进行统计,所以有可能over-represented reads不在里面。而且大于75bp的reads也是只取50bp。如果命令行中加入了-c contaminant file,出现的over-represented sequence会从contaminant_file里面找匹配的hit(至少20bp且最多一个mismatch),可以给我们一些线索。
3.10. 接头含量

本文内容由网友自发贡献,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系:hwhale#tublm.com(使用前将#替换为@)