为什么要比对:https://www.jianshu.com/p/681e02e7f9af
Jimmy老师主要演示了四种比对工具,分别为hisat2、subjunc、bowtie2、bwa。除了subjunc能够直接生成bam文件外,这些软件的用法都很相似。需要根据自己的需求来选择对应的软件。
这里以使用hisat2为例。
1. 下载对应的index
为什么要下载index:https://www.jianshu.com/p/681e02e7f9af
Hisat2官网下载:https://daehwankimlab.github.io/hisat2/download/
挂了梯子,使用edge浏览器,一小时左右下完。之后再导入服务器,再花一小时,淦!!!
解压文件
tar -zxvf hg19_genome.tar.gz
tar -zxvf mm10_genome.tar.gz
看一下里面都有什么
2. 序列比对
# 激活环境
conda activate rna
# -p 设置线程
# -x 参考基因组索引文件的前缀
# -U 单端测序文件
# -S 指定输出文件
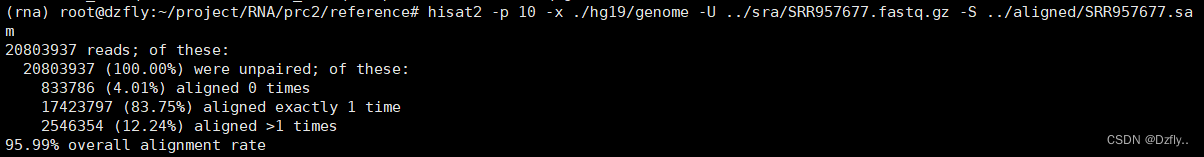
hisat2 -p 10 -x ./hg19/genome -U ../sra/SRR957677.fastq.gz -S ../aligned/SRR957677.sam
一次比对多个文件的例子:https://www.jianshu.com/p/479c7b576e6f
# 双端测序数据
# -t 显示比对时间
# -1 双端测序结果的第一个文件
# -2 双端测序结果的第二个文件
# 人的比对
for ((i=56;i<=58;i++));do hisat2 -t -x /mnt/f/rna_seq/data/reference/index/hg19/genome -1 /mnt/f/rna_seq/data/SRR35899${i}.sra_1.fastq.gz -2 /mnt/f/rna_seq/data/SRR35899${i}.sra_2.fastq.gz -S SRR35899${i}.sam ;done
# 小鼠比对
$ for ((i=59;i<=62;i++));do hisat2 -t -x /mnt/f/rna_seq/data/reference/index/mm10/genome -1 /mnt/f/rna_seq/data/SRR35899${i}.sra_1.fastq.gz -2 /mnt/f/rna_seq/data/SRR35899${i}.sra_2.fastq.gz -S SRR35899${i}.sam; done
3. samtools:将sam文件转为bam文件
sort 默认按照染色体位置进行排序
-n 根据read名进行排序
-t 根据TAG进行排序
参考:https://www.jianshu.com/p/681e02e7f9af
详细参数解释见:使用SAMtools将SAM文件转换为BAM文件、排序、建立索引
# -O 设置最终输出的文件格式,可以是bam,sam或者cram,默认为bam
# -@ 设置线程,加快运行速度
# -o 设置最终排序后的输出文件名
samtools sort -O bam -@ 2 -o SRR957677.bam SRR957677.sam
# 必须对bam文件进行默认情况下的排序后,才能进行index,否则会报错。
# 建立索引后将产生后缀为.bai的文件,用于快速的随机处理。
samtools index SRR957677.bam
samtools view SRR957677.bam | less -SN
# 给出BAM文件的比对结果
samtools flagstat -@ 2 SRR957677.bam
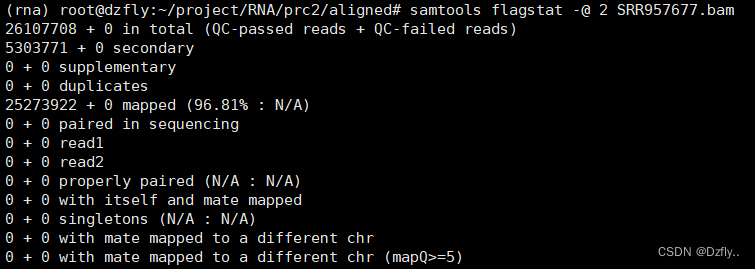
这里比对结果异常,应该是使用的数据为单端测序数据造成的。
一次转换多个文件的例子:https://www.jianshu.com/p/479c7b576e6f
# 首先将比对后的sam文件转换成bam文件
# 利用的是samtools的view选项,参数-S 输入sam文件;参数-b 指定输出的文件为bam;最后重定向写入bam文件
$ cd mnt/f/rna_seq/aligned
$ for ((i=56;i<=62;i++));do samtools view -S SRR35899${i}.sam -b > SRR35899${i}.bam;done
# 将所有的bam文件按默认的染色体位置进行排序
$ for ((i=56;i<=62;i++));do samtools sort SRR35899${i}.bam -o SRR35899${i}_sorted.bam;done
# 将所有的排序文件建立索引,索引文件.bai后缀
$ for ((i=56;i<=62;i++));do samtools index SRR35899${i}_sorted.bam;done
Jimmy老师的方法
ls *.sam | while read id; do (samtools sort -O bam -@ 5 -o $(basename ${id} ".sam").bam ${id}); done
ls *.bam | xargs -i samtools index {}
ls *.bam | while read id; do (samtools flagstat -@ 10 $id > $(basename ${id} ".bam").flagstat); done
4. 将bam文件载入IGV
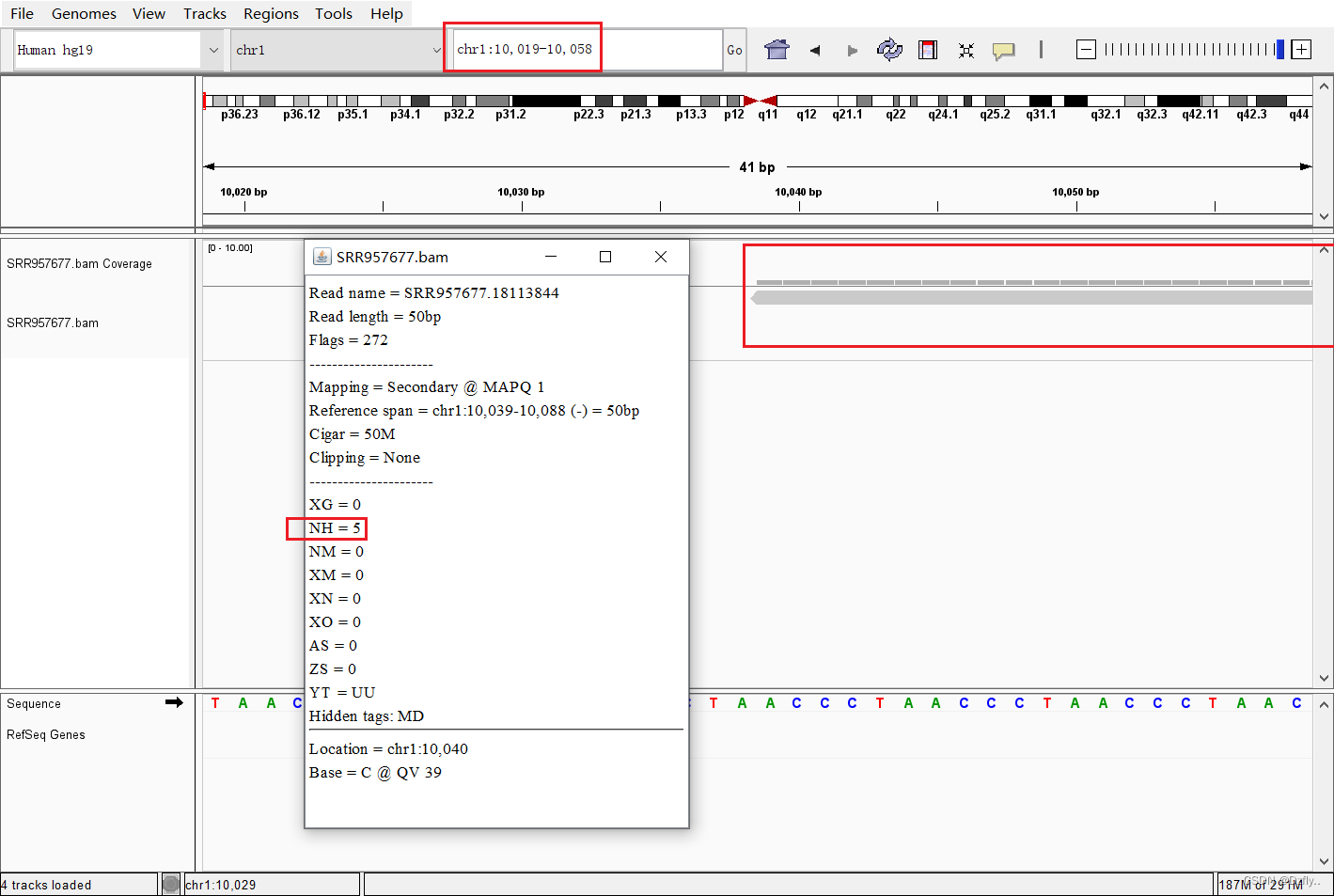
将bam文件导入igv,在chr位置输入":10039",结果如图。看不懂…